Биоинформатики избавились от лишнего шага в анализе стабильности белков
Исследователи из Центра молекулярной и клеточной биологии Сколтеха сравнили, как разные методы предсказания структуры белка влияют на дальнейшую оценку его стабильности после мутации. Оказалось, что одинаковый результат дают как экспериментально полученная трехмерная структура близкого по последовательности белка, так и предсказанная искусственным интеллектом структура исследуемого белка.
А вот попытка предсказать структуру интересующего ученых белка на основе известной строения его «родственника» только ухудшает предсказание. Это упростит исследователям предварительные вычисления при оценке изменения стабильности вследствие мутаций.
Статья с результатами исследования опубликована в журнале Bioinformatics.
В биологических экспериментах ученые часто имеют дело с мутантными белками: например, при исследовании их структуры и функций, изучении биологических процессов в клетках, белковой инженерии. Известно, что мутации могут изменять структуру и влиять на их стабильность. Поскольку эксперимент — очень трудоемкий и дорогостоящий процесс, ученые разрабатывают вычислительные методы для оценки влияний мутаций на стабильность. Однако для их применения необходимо знать трехмерную структуру белка.
Далеко не для всех белков трехмерная структура определена экспериментально, и велика вероятность, что для интересующего исследователей белка структура еще не известна. В таком случае на помощь могут прийти доступные 3D-модели «ближайших родственников» — гомологов — этого белка, поскольку известно, при какой степени сходства аминокислотной последовательности трехмерная структура белков также будет хорошо совпадать. Решение — сначала предсказать структуру исследуемого белка, взяв за основу известную структуру его гомолога, а потом рассчитать влияние мутаций для такой предсказанной модели.
В 2021 году в области предсказания структуры белков случился прорыв, и у ученых появилась альтернатива. Вместо предсказания трехмерной структуры по гомологии стало возможно воспользоваться программой на основе искусственного интеллекта AlphaFold, которая предсказывает структуру белка «с нуля» — по аминокислотной последовательности, и уже «справилась» с подавляющим большинством известных на сегодняшний день белков.
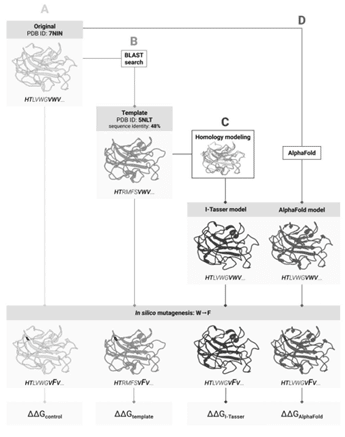 Четыре пути предсказания изменения стабильности белка после мутации: (А) по структуре исходного белка; (B) по структуре его гомолога; © по структуре исходного белка, предсказанной на основе структуры голомолга, и (D) по структуре, предсказанной искусственным интеллектом на основе аминокислотной последовательности / ©Пресс-служба Сколтеха
Четыре пути предсказания изменения стабильности белка после мутации: (А) по структуре исходного белка; (B) по структуре его гомолога; © по структуре исходного белка, предсказанной на основе структуры голомолга, и (D) по структуре, предсказанной искусственным интеллектом на основе аминокислотной последовательности / ©Пресс-служба Сколтеха
В новой работе ученые из Сколтеха задались вопросом, какой из этих подходов работает лучше для предсказаний изменений стабильности вследствие мутаций? Несмотря на высокую степень достоверности предсказаний AlphaFold, экспериментальное определение структуры белка остается «золотым стандартом».
Чтобы сравнить оба подхода, исследователи воспользовались семью различными методами для оценки изменения стабильности и сравнили их показатели для AlphaFold и I-Tasser — лучшего алгоритма предсказания структуры по гомологии. Дополнительно биологи проверили, можно ли пропустить шаг с предсказанием структуры по гомологии — и проводить расчеты стабильности для известной структуры гомологичного белка.
«Мы решили выяснить, насколько тот факт, что мы взяли не реальную структуру белка, а “соседнюю”, отклоняет нас от правильного предсказания изменений стабильности. И оказалось, что дополнительный шаг — предсказание структуры белка по гомологии — только ухудшает результат. Мы показали, что фактически нет разницы между тем, использовать ли определенную экспериментально структуру гомолога или предсказанную искусственным интеллектом AlphaFold структуру исследуемого белка. Отчасти это валидационная работа: вот, у ученых появились новые инструменты, но можно ли ими пользоваться? Сначала нужно проверить, действительно ли этот инструмент хорошо работает для нашей задачи. Такой анализ мы и провели», — рассказывает Марина Пак, аспирантка Сколтеха и первый автор исследования.
«Из-за ажиотажа вокруг успехов AlphaFold некоторые ученые и неспециалисты считают, что сейчас любая задача вычислительной биологии, касающаяся исследования белков, тем самым решена. Но это не так. Скажем, предсказание изменения стабильности вследствие мутации остается на очень невысоком уровне достоверности. При этом изменение стабильности — один из ключевых параметров, который влияет на уровень активности белка. И если бы был инструмент, который однозначно говорит о том, как изменится стабильность белка из-за мутации — это помогло бы и при планировании эксперимента, и при интерпретации результатов. Допустим, у нас есть белок, который не оптимален по стабильности. И мы хотим найти мутации, которые стабилизируют его под желаемые условия — например, чтобы он оставался активным при высокой температуре. Когда мы сможем делать это полностью вычислительно, подход к редизайну и оптимизации белков кардинально изменится», — заключает Дмитрий Иванков, старший преподаватель Сколтеха и руководитель исследования.
Несмотря на то, что задача предсказания изменения стабильности выглядит проще, чем предсказание трехмерной структуры, пока она не под силу даже искусственному интеллекту. Одна из проблем — ограниченный объем данных для обучения: в то время как в распоряжении AlphaFold было почти две сотни тысяч белковых структур для обучения, экспериментальные данные по изменению стабильности измеряются тысячами, причем относятся они всего к нескольким десяткам уникальных белков. С увеличением объема данных и повышением интереса исследователей к этой задаче в ближайшем будущем удастся совершить прорыв, надеются авторы работы.
- Источник(и):
- Войдите на сайт для отправки комментариев
