О некоторых возможностях современного квантово-механического ПО
Известно, что прогресс в вычислительной технике в период последних 5-ти лет позволил выйти квантово-механическому моделированию из суперкомпьютерных центров к «широкому кругу» пользователей путем повсеместного внедрения в университетах и на производстве недорогих, но высокопроизводительных вычислительных кластеров. Квантово-механическое ПО развивается не менее быстрым темпом.
Рассмотрим некоторый возможности современного ПО для проведения квантово-механических расчетов при моделировании наноструктур.
Наиболее качественные результаты моделирования возможны при использовании ПО, реализующего методы теории функционала плотности (ТФП), или ab-initio методы (моделирование «из первых принципов»). Результаты моделирования, приведенные в этой статье получены на условно-бесплатном ПО, использующем ТФП (англ. DFT): AbInit, Siesta, Dacapo, Exciting, Fleur, Elk, FHImd.
Молекулярная динамика наноструктур. Молекулярная динамика – подход, при котором моделируют движение каждого атома в молекулярной системе для того, чтобы наблюдать кинетическое поведение системы и ее свойства в равновесном состоянии при заданных параметрах окружающей среды.
 Кристалл меди при Т=0 К
Кристалл меди при Т=0 К
 Кристалл меди (распад) при Т=2000 К
Кристалл меди (распад) при Т=2000 К
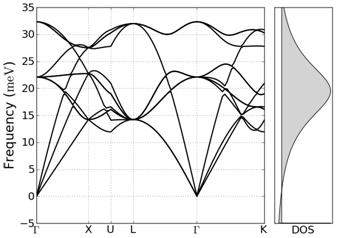 Фононный спектр кристалла меди
Фононный спектр кристалла меди
Прочностные расчеты наноструктур путем построения энергетических зависимостей для последующего определения модуля упругости.
Предлагается добиться упрочнения покрытий из нитрида и карбида титана введением примесей порядка 1–2 % .
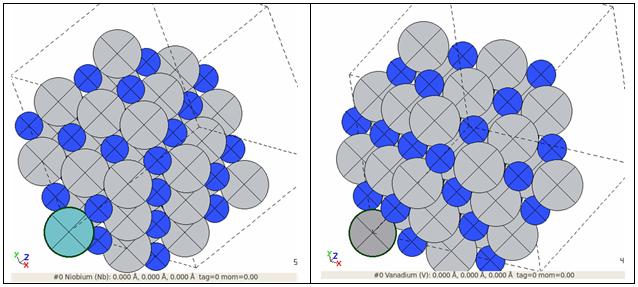 Введение атома примеси ниобия (слева) и ванадия (справа) в супер-ячейку массива нитрида титана
Введение атома примеси ниобия (слева) и ванадия (справа) в супер-ячейку массива нитрида титана
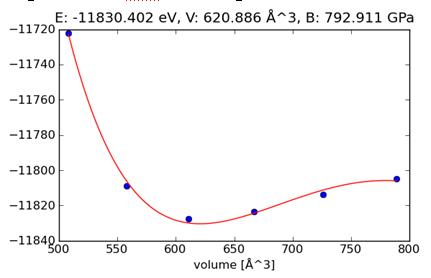 Построение энергетической зависимости супер-ячейки объемного нитрида титана от ее размера, для нахождения модуля упругости по методу Бирч-Мурнагана, примесь Nb, модуль упругости B = 793.
Построение энергетической зависимости супер-ячейки объемного нитрида титана от ее размера, для нахождения модуля упругости по методу Бирч-Мурнагана, примесь Nb, модуль упругости B = 793.
Упрочнение примесями переходных металлов показало достаточно высокую эффективность, в особенности танталом Ta (прочность возросла на 29% для TiN и на 42% для TiC) и ниобием Nb (прочность возросла на 28% для TiN и на 41% для TiC).
Определение устойчивых конфигураций интерметаллидов путем построения фазовых диаграмм.
Фазой называется часть или совокупность гомогенных частей интерметаллида, разделённых границами раздела, обладающие одинаковым составом, структурой, свойствами и при переходе через границу раздела структуры, состав, свойства или структура могут меняться скачкообразно.
Графическим способом описания фаз, находящихся в состоянии равновесия, является диаграмма состояния.
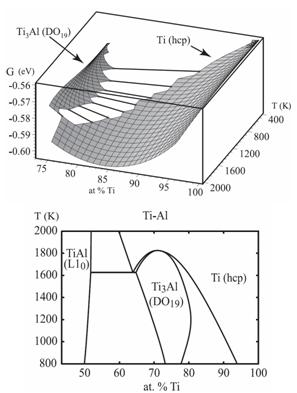 Фазовые диаграммы для интерметаллида TiAl в 3D и 2D
Фазовые диаграммы для интерметаллида TiAl в 3D и 2D
Получено пространство состояний интерметаллидов семейства TiAl, широко применяемых в качестве упрочняющих покрытий. Выявлены устойчивые состояния конфигураций интерметаллидов на диапазоне температур и концентраций путем расчета и построения фазовых диаграмм и распределений энергии.
Расчет распределения электронной плотности (англ. DOS) и зонной структуры (англ. BAND) вещества.
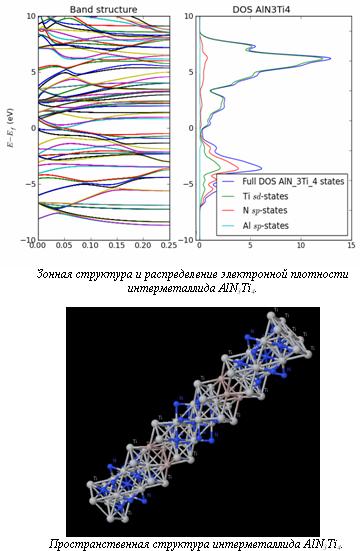
Расчет распределения электронной плотности и зонной структуры необходим для исследования электродинамических свойств материалов. Расчеты разрешенных и запрещенных зон, значений электронной плотности на уровне Ферми используются для анализа электронных свойств в основном для полупроводниковых материалов и диэлектриков, изучения явления сверхпроводимости.
Исследование взаимодействий материалов на границе сред.
Прочность сцепления материалов между собой (подложки и покрытия) определяется величиной энергетического барьера перемещения атома покрытия из текущей ячейки, состоящую из атомов подложки, в соседнюю ячейку кристаллической решетки.
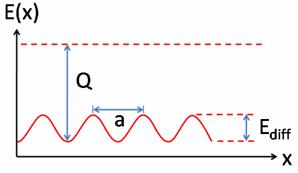 Энергетический ландшафт поверхности наноструктуры при диффузии на ней атомов в одном измерении (Х – ось перемещения атомов по поверхности подложки), Q-энергия связи взаимодействий на границе сред, Ediff является барьером для диффузии, a – расстояние между соседними узлами адсорбции
Энергетический ландшафт поверхности наноструктуры при диффузии на ней атомов в одном измерении (Х – ось перемещения атомов по поверхности подложки), Q-энергия связи взаимодействий на границе сред, Ediff является барьером для диффузии, a – расстояние между соседними узлами адсорбции
Миграция материала покрытия (титан, в данном исследовании) не является свободной вследствие наличия энергетических барьеров, которые имеются на поверхности подложки с правильной кристаллической решеткой (гексагональной в случае кобальта, в данном исследовании). При диффузии (адсорбции) на границе кристаллов возникает энергетическая неоднородность вследствие периодичности в расположении элементов кристаллической решетки, которая препятствует этим процессам пропорционально энергетическому барьеру, который можно рассчитать, используя методы квантово-механической оптимизации (применяется метод «QuasiNewton»).
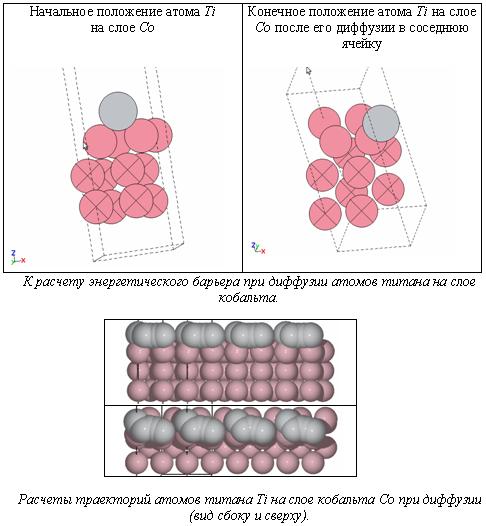
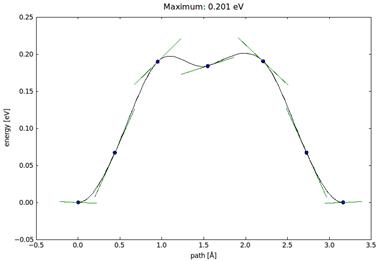 Энергетический барьер (Ediff=0.201 eV) для атома титана на поверхности кобальта
Энергетический барьер (Ediff=0.201 eV) для атома титана на поверхности кобальта
По величине энергетического барьера (Ebarr= 0.201 eV для преодоления расстояния около 3A) следует судить о прочности позиционирования покрытия на подложке. При условии достаточно высокого барьера какая-либо динамика на границе материалов затруднительна, в том числе при внешних механических и температурных воздействиях.
Продолжение следует…
Автор: Сергей В. Серый,
к. т. н. , доцент, докторант, специальность 05.13.18 – «Мат. моделирование, численные методы и комплексы программ», нач. отдела «Математического моделирования динамических систем и наноструктур» ФГБОУ ВПО КнАГТУ.
- Войдите на сайт для отправки комментариев
